Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis, is an autoimmune disease of the liver.[1][2][3] It results from a slow, progressive destruction of the small bile ducts of the liver, causing bile and other toxins to build up in the liver, a condition called cholestasis. Further slow damage to the liver tissue can lead to scarring, fibrosis, and eventually cirrhosis.
| Primary biliary cholangitis | |
|---|---|
| Other names | Primary biliary cirrhosis |
 | |
| Micrograph of PBC showing bile duct inflammation and injury, H&E stain | |
| Specialty | Gastroenterology, Hepatology |
| Symptoms | Cholestasis, pruritus, fatigue |
| Complications | Cirrhosis, hepatic failure, portal hypertension |
| Usual onset | Usually middle-aged women |
| Causes | Autoimmune |
| Risk factors | Female sex |
| Diagnostic method | Anti-mitochondrial antibodies, Liver biopsy |
| Differential diagnosis | Autoimmune hepatitis |
| Treatment | Ursodeoxycholic acid, obeticholic acid, cholestyramine |
| Frequency | 1 in 3,000–4,000 people |
Common symptoms are tiredness, itching, and in more advanced cases, jaundice. In early cases, the only changes may be those seen in blood tests.[4]
PBC is a relatively rare disease, affecting up to one in 3,000–4,000 people.[5][6] As with many other autoimmune diseases, it is much more common in women,[7] with a sex ratio of at least 9:1 female to male.[1] The reasons for this disparity are unclear, but may involve the expression of sex hormones such as estrogen, which impact immune system response.[7]
The condition has been recognised since at least 1851, and was named "primary biliary cirrhosis" in 1949.[8] Because cirrhosis is a feature only of advanced disease, a change of its name to "primary biliary cholangitis" was proposed by patient advocacy groups in 2014.[9][10]
Signs and symptoms
editPeople with PBC experience fatigue (80%); this is a nonspecific symptom and can be debilitating, with a huge impact on quality of life. Its pathogenesis is still unknown, and is quite challenging to explore its specificity and to treat. Comorbidities that could contribute to or worsen fatigue, such as depression, hypothyroidism, anaemia, obesity, or medication side effects, should be promptly identified and treated. Dry skin and dry eyes are also common. Itching (pruritus) occurs in 20–70% of cases,[4] and can develop at any stage of the disease. Textbooks tend to describe itching in the feet and hands, but patients may also experience itching of the scalp, face, back, or other areas. The itching is typically mild-to-moderate in intensity. It may manifest as a tingling, crawling or burning sensation, and can develop even with normal liver function tests. It does not correlate with progression of liver disease, and may even improve or disappear as the disease advances. Given the impact on quality of life and night sleep, pruritus is also correlated with fatigue. It can rarely be severe, non-responsive to medical therapy, and requiring liver transplant. Pruritus is characteristically intermittent, worse at night, and improves during summer. People with more severe PBC may have jaundice (yellowing of the eyes and skin).[4] PBC impairs bone density and the risk of fracture increases.[4] Xanthelasma (skin lesions around the eyes) or other xanthoma may be present as a result of increased cholesterol levels.[11]
PBC can eventually progress to cirrhosis of the liver. This, in turn, may lead to a number of symptoms or complications, including:
- Fluid retention in the abdomen (ascites) in more advanced disease
- Enlarged spleen in more advanced disease
- Oesophageal varices in more advanced disease
- Hepatic encephalopathy, including coma in extreme cases in more advanced disease.
People with PBC may also sometimes have the findings of an associated extrahepatic autoimmune disorder such as thyroid disease or rheumatoid arthritis or Sjögren's syndrome (in up to 80% of cases).[11][12]
Causes
editPBC has an immunological basis, and is classified as an autoimmune disorder. It results from a slow, progressive destruction of the small bile ducts of the liver, with the intralobular ducts and the canals of Hering (intrahepatic ductules) being affected early in the disease.
Most people with PBC (more than 90%) have antimitochondrial antibodies (AMAs) against pyruvate dehydrogenase complex (PDC-E2), an enzyme complex found in the mitochondria. People who are negative for AMAs are usually found to be positive when more sensitive methods of detection are used.[13]
People with PBC may also have been diagnosed with another autoimmune disease, such as a rheumatological, endocrinological, gastrointestinal, pulmonary, or dermatological condition, suggesting shared genetic and immune abnormalities.[12] Common associations include Sjögren's syndrome, systemic sclerosis, rheumatoid arthritis, lupus, hypothyroidism, and coeliac disease.[14][15]
A genetic predisposition to disease has been thought to be important for some time. Evidence for this includes cases of PBC in family members, identical twins both having the condition (concordance), and clustering of PBC with other autoimmune diseases. In 2009, a Canadian-led group of investigators reported in the New England Journal of Medicine results from the first PBC genome-wide association study.[16][17] This research revealed parts of the IL12 signaling cascade, particularly IL12A and IL12RB2 polymorphisms, to be important in the aetiology of the disease in addition to the HLA region. In 2012, two independent PBC association studies increased the total number of genomic regions associated to 26, implicating many genes involved in cytokine regulation such as TYK2, SH2B3 and TNFSF11.[18][19]
A study of over 2,000 patients identified a gene, POGLUT1, that appeared to be associated with this condition.[20] Earlier studies have also suggested that this gene may be involved. The implicated protein is an endoplasmic reticulum O-glucosyltransferase.
An environmental Gram-negative Alphaproteobacterium — Novosphingobium aromaticivorans[21] has been associated with this disease, with several reports suggesting an aetiological role for this organism.[22][23][24] The mechanism appears to be a cross-reaction between the proteins of the bacterium and the mitochondrial proteins of the liver cells. The gene encoding CD101 may also play a role in host susceptibility to this disease.[25]
A failure of immune tolerance against the mitochondrial pyruvate dehydrogenase complex (PDC-E2) is a primary cause, with shedding of the antigen into apoptotic bodies or "apotopes" leading to the anatomic localization.[26] Such autoreactivity may also be the case with other proteins, including the gp210 and p62 nuclear pore proteins. Gp210 has increased expression in the bile duct of anti-gp210 positive patients, and these proteins may be associated with prognosis.[27]
Diagnosis
editMost patients are currently diagnosed when asymptomatic, having been referred to the hepatologist for abnormal liver function tests (mostly raised GGT or alkaline phosphatase) performed for annual screening blood tests. Other frequent scenarios include screening of patients with nonliver autoimmune diseases, e.g. rheumatoid arthritis, or investigation of elevated cholesterol, evaluation of itch or unresolved cholestasis post partum. Diagnosing PBC is generally straightforward. The basis for a definite diagnosis are:
- Abnormalities in liver enzyme tests are usually present and elevated gamma-glutamyl transferase and alkaline phosphatase are found in early disease.[11] Elevations in bilirubin occur in advanced disease.
- Antimitochondrial antibodies are the characteristic serological marker for PBC, being found in 90–95% of patients and only 1% of controls. PBC patients have AMA against pyruvate dehydrogenase complex (PDC-E2), an enzyme complex that is found in the mitochondria.[11] Those people who are AMA negative but with disease similar to PBC have been found to have AMAs when more sensitive detection methods are employed.[13]
- Other auto-antibodies may be present:
- Antinuclear antibody measurements are not diagnostic for PBC because they are not specific, but may have a role in prognosis.
- Anti-glycoprotein-210 antibodies, and to a lesser degree anti-p62 antibodies, correlate with the disease's progression toward end-stage liver failure. Anti-gp210 antibodies are found in 47% of PBC patients.[28][29]
- Anti-centromere antibodies often correlate with developing portal hypertension.[30]
- Anti-np62[31] and anti-sp100 are also found in association with PBC.
- Abdominal ultrasound, magnetic resonance cholangiopancreatography or a CT scan is usually performed to rule out blockage to the bile ducts. This may be needed if a condition causing secondary biliary cirrhosis, such as other biliary duct disease or gallstones, needs to be excluded. A liver biopsy may help, and if uncertainty remains as in some patients, an endoscopic retrograde cholangiopancreatography, an endoscopic investigation of the bile duct, may be performed.
Given the high specificity of serological markers, liver biopsy is not necessary for the diagnosis of PBC; however, it is still necessary when PBC-specific antibodies are absent, or when co-existent autoimmune hepatitis or nonalcoholic steatohepatitis is suspected. Liver biopsy can be useful to stage the disease for fibrosis and ductopenia. Finally, it may also be appropriate in the presence of other extrahepatic comorbidities.
-
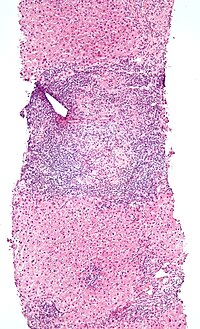 Low-magnification micrograph of PBC, H&E stain
Low-magnification micrograph of PBC, H&E stain -
 Intermediate-magnification micrograph of PBC showing bile duct inflammation and periductal granulomas, liver biopsy, H&E stain
Intermediate-magnification micrograph of PBC showing bile duct inflammation and periductal granulomas, liver biopsy, H&E stain -
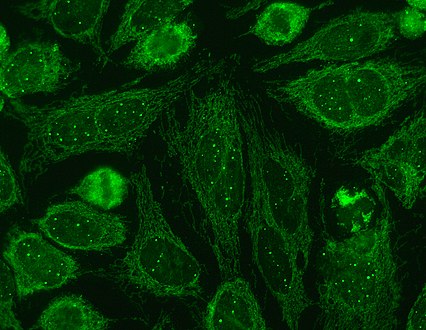 Immunofluorescence staining pattern of sp100 antibodies (nuclear dots) and antimitochondrial antibodies
Immunofluorescence staining pattern of sp100 antibodies (nuclear dots) and antimitochondrial antibodies



Liver biopsy
editOn microscopic examination of liver biopsy specimens, PBC is characterized by chronic, nonsuppurative inflammation, which surrounds and destroys interlobular and septal bile ducts. These histopathologic findings in primary biliary cholangitis include:[32]
- Inflammation of the bile ducts, characterized by intraepithelial lymphocytes
- Periductal epithelioid granulomas.
- Proliferation of bile ductules
- Fibrosis (scarring)
The Ludwig and Scheuer scoring systems have historically been used to stratify four stages of PBC, with stage 4 indicating the presence of cirrhosis. In the new system of Nakanuma, the stage of disease is based on fibrosis, bile duct loss, and features of cholestasis, i.e. deposition of orcein-positive granules, whereas the grade of necroinflammatory activity is based on cholangitis and interface hepatitis. The accumulation of orcein-positive granules occurs evenly across the PBC liver, which means that staging using the Nakanuma system is more reliable regarding sampling variability.
Liver biopsy for the diagnosis and staging of PBC lost favour after the evidence of a patchy distribution of the duct lesions and fibrosis across the organ. The widespread availability of noninvasive measures of fibrosis means that liver biopsy for staging of PBC is somewhat obsolete. Liver biopsy does, however, remain useful in certain settings. The main indications are to confirm the diagnosis of PBC when PBC-specific antibodies are absent and confirm a diagnosis of PBC with AIH features (i.e. overlap PBC-AIH). Liver biopsy is also useful to assess the relative contribution of each liver injury when a comorbid liver disease is present, such as non-alcoholic steatohepatitis. In patients with inadequate response to UDCA, liver biopsy may provide the explanation and could undoubtedly inform risk stratification. For example, it may identify a previously unsuspected variant syndrome, steatohepatitis, or interface hepatitis of moderate or greater severity. It is also useful in AMA and ANA-specific antibody negative cholestatic patients to indicate an alternative process, e.g. sarcoidosis, small duct PSC, adult idiopathic ductopenia.
Histopathology stages (by Ludwig and Scheuer systems)
edit- Stage 1 – portal stage: Normal-sized triads, portal inflammation, subtle bile duct damage: Granulomas are often detected in this stage.
- Stage 2 – periportal stage: Enlarged triads, periportal fibrosis and/or inflammation, typically characterized by the finding of a proliferation of small bile ducts
- Stage 3 – septal stage: Active and/or passive fibrous septa
- Stage 4 – biliary cirrhosis: Nodules present, garland or jigsaw puzzle pattern
Treatment
editCholestasis
editMedical therapy of PBC targets disease progression and symptom control. The first-line treatment for PBC is ursodeoxycholic acid (UDCA).[33][34] UDCA has been the only drug available for two decades and more recently obeticholic acid (OCA), a semi-synthetic hydrophobic bile acid analogue, has been licensed in patients failing UDCA response or intolerant to UDCA. Several other agents have been studied, including immunosuppressants, but robust evidence of benefit is lacking.[11][35][36]
UDCA improves liver enzyme levels, slows down histological progression, and improves liver transplant-free survival.[37][11] UDCA also reduces the need for liver transplantation.[33] UDCA should be taken at a dose of 13 to 15 mg per kg of body weight per day,[34] usually in two divided doses each day.[33] Liver chemistries usually improve within a few weeks of starting UDCA, and 90% of any benefit is observed after 6–9 months of therapy.[33] Liver chemistries should be re-evaluated after 1 year of treatment.[33] UDCA is usually continued lifelong.[34] Up to 40% of people do not respond to treatment with UDCA.[33] Patients with PBC who have an inadequate response to UDCA or those few (less than 3%) who are intolerant to UDCA are candidates for second-line therapies.
Obeticholic acid (OCA) is FDA-approved for the treatment of PBC in individuals intolerant or unresponsive to UDCA.[33] OCA is a farnesoid X receptor agonist, and results in increased bile flow (choleresis). OCA is started at 5 mg daily, and liver chemistries should be rechecked after 3 months of treatment. If the liver chemistries remain elevated, then the dose of OCA may be increased to 10 mg per day. The most common side effect of OCA is pruritus.
Fibric acid derivatives, or fibrates, are agonists of the peroxisome proliferator activator receptor (PPAR), a nuclear receptor involved in several metabolic pathways. While fibrates are approved for the treatment of hypertriglyceridemia, they exert anticholestatic effects and have been studied for the treatment of PBC.[33] Among the fibrates, bezafibrate and fenofibrate, PPAR-alpha selective agonists, have been extensively studied as therapeutic agents because of their potential ability to decrease bile acid synthesis and bile acid-related hepatic inflammation. A randomized, controlled trial in 2018 showed its efficacy in patients with inadequate response to UDCA. While fibrates can be considered as off-label treatment for PBC that does not respond to UDCA, they should not be used in decompensated cirrhosis.[33]
Elafibranor (Iqirvo) was approved for medical use in the United States in June 2024.[38]
Additional medications are being investigated as potential treatments for PBC, and found to be ineffective as single agents (monotherapy), including: chlorambucil, colchicine, cyclosporine, corticosteroids, azathioprine, malotilate, methotrexate, mycophenolate mofetil, penicillamine, and thalidomide.[33] Budesonide may be used as an off-label treatment for PBC, although its efficacy is controversial.[33] Seladelpar, a PPAR-delta receptor agonist, is being studied for treatment of PBC.[39][40]
Seladelpar (Livdelzi) was approved for medical use in the United States in August 2024.[41]
Itching
editPruritus is a common symptom in people with PBC. First-line treatment of pruritus consists of anion-exchange resins, such as cholestyramine, colestipol, or colesevalam.[33] These anion-exchange resins are nonabsorbed, highly positively charged substances that bind bile acids, which are negatively charged anions.[33] Anion-exchange resins relieve itching caused by excess bile acids in circulation by binding bile acids in the gut and facilitating elimination. Bloating or constipation may occur with anion-exchange resins.[33] Cholestyramine may affect absorption of UDCA; if cholestyramine is necessary, it should be taken at least 60 minutes before or 4 hours after UDCA is taken.[33]
Treatment options for pruritus that does not improve with anion-exchange resins include: rifampicin, naltrexone, or sertraline.[33] Rifampicin may rarely cause drug induced liver injury and should be avoided if serum bilirubin is elevated (greater than 2.5 mg/dL). Liver enzymes should be monitored after starting rifampin.[34] Rifampicin induces enzymes, resulting in numerous potential drug-drug interactions.[33] Opioid antagonists may cause a self-limited opioid withdrawal like reaction, with abdominal pain, elevated blood pressure, tachycardia, goose bumps, nightmares, and depersonalization.[33] To avoid such reactions, the dose should start low and gradually be increased.[33]
Other therapies
edit- Fatigue is a nonspecific but often-reported symptom in PBC, and represents an unmet need since no therapies are licensed. A structured approach to management, quantifying fatigue and its impacts (through the use of disease-specific tools such as the PBC-40 quality-of-life measures), addressing contributing and exacerbating factors, and supporting patients to cope with its impact is effective. Drugs such as coenzyme Q and rituximab have been shown to be ineffective. A graded programme of exercise helps some individuals.[citation needed]
- People with PBC may have poor lipid-dependent absorption of oil-soluble vitamins (A, D, E, and K).[42] Appropriate supplementation is recommended when bilirubin is elevated.[11]
- People with PBC are at elevated risk of developing osteoporosis[43] as compared to the general population and others with liver disease. Screening and treatment of this complication is an important part of the management of PBC.
- As in all liver diseases, consumption of alcohol should be restricted or eliminated.
- In patients with advanced liver disease, the only curative therapy is liver transplant. Outcomes are favourable, with five-year patient survival rates better than for most other indications for LT (80–85%).[44][45]
Prognosis
editThe introduction of UDCA has dramatically changed the pattern and the course of the disease. Numerous trials and observational studies have demonstrated its efficacy on liver biochemistry, histological progression, and transplant-free survival.[46]
Among the UDCA-treated patients, the degree of the liver biochemistry improvement, i.e. the UDCA-response, identifies patients with different long-term prognosis. In the absence of cirrhosis, people who experience an improvement of liver enzymes to the normal range on treatment with UDCA have excellent survival, which may be similar to the general population.[47] Survival is significantly reduced though, in those with abnormal liver biochemistry on treatment.
The two most important parameters in evaluating response to UDCA are alkaline phosphatase and total bilirubin. Qualitative and quantitative definitions of UDCA-response have been developed, based on changes of bilirubin, transaminases and ALP, after a period of 6 to 24 months of treatment with UDCA at 13–15 mg/kg/day.[48]
Patients at diagnosis can be risk-stratified based on the probability of UDCA-response. This is relevant to identify patients who would be eligible for second-line therapies before waiting for the treatment failure under UDCA, with potential impact on disease course.[49]
Hepatocellular carcinoma (HCC) is infrequent in PBC. Recent large-scale cohort studies highlighted that the lack of UDCA-response after 12 months of therapy and male sex are associated with increased future risk of developing HCC in PBC.
After liver transplant, the recurrence of disease may be as high as 18% at five years, and up to 30% at 10 years. No consensus exists on risk factors for recurrence of the disease.[50]
Epidemiology
editEpidemiologic studies report heterogeneous incidence rates of 0.33 to 5.8 per 100,000 inhabitants per year, and prevalence rates of 1.9 to 40.2 per 100,000 inhabitants. Such figures, in particular the prevalence, have shown some increase in the last decades. Improvement of diagnostic tools, increasing disease awareness, and digitised patient registration with facilitation of case-findings, along with improved survival, likely contributed to the rising prevalence rates. The disease has been described worldwide, though North America and Northern Europe have shown the highest incidence and prevalence rates. Whether a true variation in disease prevalence exists among populations of different geographical areas and of different ethnicity or if this is a consequence of a difference in study quality is unknown.[5][6] PBC is more common in women, with a female:male ratio of at least 9:1. The peak incidence of PBC is in the fifth decade of life. In some areas of the US and UK, the prevalence is estimated to be as high as one in 4,000. This is much more common than in South America or Africa, which may be due to better recognition in the US and UK.[5][6] First-degree relatives may have as much as a 500 times increase in prevalence, but if this risk is greater in the same-generation relatives or the one that follows is debated.
PBC is considered a prime example of the female preponderance in autoimmunity with a female to male ratio of up to 9:1, confirmed by large cohort studies, although some recent data, using administrative registries, suggest an increasing male prevalence. Major defects of sex chromosomes, i.e. enhanced monosomy X in female patients and an enhanced Y chromosome loss in male patients, have been described and might well explain the greater female predisposition to develop PBC.[51]
An association of a greater incidence of PBC at latitudes more distant from the Equator is similar to the pattern seen in multiple sclerosis.[52]
Typical disease onset is between 30 and 60 years, though cases have been reported of patients diagnosed at the ages of 15 and 93. Prevalence of PBC in women over the age of 45 years could exceed one in an estimated 800 individuals.
History
editThe first report of the disease dates back 1851 by Addison and Gull, who described a clinical picture of progressive jaundice in the absence of mechanical obstruction of the large bile ducts. Ahrens et al. in 1950 published the first detailed description of 17 patients with this condition, and coined the term "primary biliary cirrhosis". In 1959, Dame Sheila Sherlock reported a further series of PBC patients and recognised that the disease could be diagnosed in a precirrhotic stage and proposed the term "chronic intrahepatic cholestasis" as more appropriate description of this disease, but this nomenclature failed to gain acceptance, and the term "primary biliary cirrhosis" lasted for decades. In 2014, to correct the inaccuracy and remove the social stigma of cirrhosis, as well as all the misunderstanding, disadvantages, and discrimination emanating from this misnomer in daily life for patients, international liver associations agreed to rename the disease "primary biliary cholangitis", as it is now known.[53][8][54][55]
Society and culture
editSupport groups
editPBC Foundation
editThe PBC Foundation is a UK-based international charity offering support and information to people with PBC and their families and friends.[56] It campaigns for increasing recognition of the disorder, improved diagnosis, and treatments, and estimates over 8,000 people are undiagnosed in the UK.[57][58] The Foundation has supported research into PBC including the development of the PBC-40 quality of life measure published in 2004[59] and helped establish the PBC Genetics Study.[18][60] It was founded by Collette Thain in 1996, after she was diagnosed with the condition.[57] Thain was awarded an MBE Order of the British Empire in 2004 for her work with the Foundation.[61] The PBC Foundation helped initiate the name change campaign in 2014.[9][10][62]
PBCers Organization
editThe PBCers Organization is a US-based nonprofit patient support group that was founded by Linie Moore in 1996; it advocates for greater awareness of the disease and new treatments.[63] It supported the name change initiative.[10]
References
edit- ^ a b Poupon R (May 2010). "Primary biliary cirrhosis: a 2010 update". Journal of Hepatology. 52 (5): 745–758. doi:10.1016/j.jhep.2009.11.027. PMID 20347176.
- ^ Hirschfield GM, Gershwin ME (January 2013). "The immunobiology and pathophysiology of primary biliary cirrhosis". Annual Review of Pathology. 8: 303–330. doi:10.1146/annurev-pathol-020712-164014. PMID 23347352.
- ^ Dancygier H (2010). Principles and Practice of Clinical Hepatology. Springer. pp. 895–. ISBN 978-3-642-04509-7. Retrieved 29 June 2010.
- ^ a b c d Selmi C, Bowlus CL, Gershwin ME, Coppel RL (May 2011). "Primary biliary cirrhosis". Lancet. 377 (9777): 1600–1609. doi:10.1016/S0140-6736(10)61965-4. PMID 21529926. S2CID 2741153.
- ^ a b c Boonstra K, Beuers U, Ponsioen CY (May 2012). "Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review". Journal of Hepatology. 56 (5): 1181–1188. doi:10.1016/j.jhep.2011.10.025. PMID 22245904.
- ^ a b c James OF, Bhopal R, Howel D, Gray J, Burt AD, Metcalf JV (August 1999). "Primary biliary cirrhosis once rare, now common in the United Kingdom?". Hepatology. 30 (2): 390–394. doi:10.1002/hep.510300213. PMID 10421645. S2CID 25248575.
- ^ a b Moulton VR (2018). "Sex Hormones in Acquired Immunity and Autoimmune Disease". Frontiers in Immunology. 9: 2279. doi:10.3389/fimmu.2018.02279. ISSN 1664-3224. PMC 6180207. PMID 30337927.
- ^ a b Dauphinee JA, Sinclair JC (July 1949). "Primary biliary cirrhosis". Canadian Medical Association Journal. 61 (1): 1–6. PMC 1591584. PMID 18153470.
- ^ a b PBC Foundation (UK). "PBC Name Change". Retrieved 27 January 2017.
- ^ a b c "Primary Biliary Cirrhosis Name Change Initiative" (PDF). PBCers Organization. Archived from the original (PDF) on 19 April 2015. Retrieved 15 September 2022.
- ^ a b c d e f g Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ (July 2009). "Primary biliary cirrhosis". Hepatology. 50 (1): 291–308. doi:10.1002/hep.22906. PMID 19554543. S2CID 439839.
The AASLD Practice Guideline
- ^ a b Floreani A, Franceschet I, Cazzagon N (August 2014). "Primary biliary cirrhosis: overlaps with other autoimmune disorders". Seminars in Liver Disease. 34 (3): 352–360. doi:10.1055/s-0034-1383734. hdl:11577/3143751. PMID 25057958. S2CID 21382956.
- ^ a b Vierling JM (February 2004). "Primary Biliary Cirrhosis and Autoimmune Cholangiopathy". Clinics in Liver Disease. 8 (1): 177–194. doi:10.1016/S1089-3261(03)00132-6. PMID 15062200.
- ^ Narciso-Schiavon JL, Schiavon LL (February 2017). "To screen or not to screen? Celiac antibodies in liver diseases". World Journal of Gastroenterology (Review). 23 (5): 776–791. doi:10.3748/wjg.v23.i5.776. PMC 5296194. PMID 28223722.

- ^ Volta U, Rodrigo L, Granito A, Petrolini N, Muratori P, Muratori L, et al. (October 2002). "Celiac disease in autoimmune cholestatic liver disorders". The American Journal of Gastroenterology. 97 (10): 2609–2613. doi:10.1111/j.1572-0241.2002.06031.x. PMID 12385447. S2CID 17375127.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Lu Y, et al. (June 2009). "Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants". The New England Journal of Medicine. 360 (24): 2544–2555. doi:10.1056/NEJMoa0810440. PMC 2857316. PMID 19458352.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ "UK-PBC – Stratified Medicine in Primary Biliary Cholangitis (PBC; formally known as Cirrhosis)".
- ^ a b Liu JZ, Almarri MA, Gaffney DJ, Mells GF, Jostins L, Cordell HJ, et al. (October 2012). "Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis". Nature Genetics. 44 (10): 1137–1141. doi:10.1038/ng.2395. PMC 3459817. PMID 22961000.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Juran BD, Hirschfield GM, Invernizzi P, Atkinson EJ, Li Y, Xie G, et al. (December 2012). "Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants". Human Molecular Genetics. 21 (23): 5209–5221. doi:10.1093/hmg/dds359. PMC 3490520. PMID 22936693.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Hitomi Y, Ueno K, Kawai Y, Nishida N, Kojima K, Kawashima M, et al. (January 2019). "POGLUT1, the putative effector gene driven by rs2293370 in primary biliary cholangitis susceptibility locus chromosome 3q13.33". Scientific Reports. 9 (1): 102. Bibcode:2019NatSR...9..102H. doi:10.1038/s41598-018-36490-1. PMC 6331557. PMID 30643196.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Selmi C, Balkwill DL, Invernizzi P, Ansari AA, Coppel RL, Podda M, et al. (November 2003). "Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium". Hepatology. 38 (5): 1250–1257. doi:10.1053/jhep.2003.50446. PMID 14578864. S2CID 22691658.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Mohammed JP, Mattner J (July 2009). "Autoimmune disease triggered by infection with alphaproteobacteria". Expert Review of Clinical Immunology. 5 (4): 369–379. doi:10.1586/ECI.09.23. PMC 2742979. PMID 20161124.
- ^ Kaplan MM (November 2004). "Novosphingobium aromaticivorans: a potential initiator of primary biliary cirrhosis". The American Journal of Gastroenterology. 99 (11): 2147–2149. doi:10.1111/j.1572-0241.2004.41121.x. PMID 15554995. S2CID 19177405.
- ^ Selmi C, Gershwin ME (July 2004). "Bacteria and human autoimmunity: the case of primary biliary cirrhosis". Current Opinion in Rheumatology. 16 (4): 406–410. doi:10.1097/01.bor.0000130538.76808.c2. PMID 15201604. S2CID 23532625.
- ^ Mohammed JP, Fusakio ME, Rainbow DB, Moule C, Fraser HI, Clark J, et al. (July 2011). "Identification of Cd101 as a susceptibility gene for Novosphingobium aromaticivorans-induced liver autoimmunity". Journal of Immunology. 187 (1): 337–349. doi:10.4049/jimmunol.1003525. PMC 3134939. PMID 21613619.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Lleo A, Selmi C, Invernizzi P, Podda M, Coppel RL, Mackay IR, et al. (March 2009). "Apotopes and the biliary specificity of primary biliary cirrhosis". Hepatology. 49 (3): 871–879. doi:10.1002/hep.22736. PMC 2665925. PMID 19185000.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Nakamura M, Takii Y, Ito M, Komori A, Yokoyama T, Shimizu-Yoshida Y, et al. (March 2006). "Increased expression of nuclear envelope gp210 antigen in small bile ducts in primary biliary cirrhosis". Journal of Autoimmunity. 26 (2): 138–145. doi:10.1016/j.jaut.2005.10.007. PMID 16337775.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Nickowitz RE, Worman HJ (December 1993). "Autoantibodies From Patients With Primary Biliary Cirrhosis Recognize a Restricted Region Within the Cytoplasmic Tail of Nuclear Pore Membrane Glycoprotein Gp210". The Journal of Experimental Medicine. 178 (6): 2237–2242. doi:10.1084/jem.178.6.2237. PMC 2191303. PMID 7504063.
- ^ Bauer A, Habior A (2007). "Measurement of gp210 autoantibodies in sera of patients with primary biliary cirrhosis". Journal of Clinical Laboratory Analysis. 21 (4): 227–231. doi:10.1002/jcla.20170. PMC 6648998. PMID 17621358.
- ^ Nakamura M, Kondo H, Mori T, Komori A, Matsuyama M, Ito M, et al. (January 2007). "Anti-gp210 and anti-centromere antibodies are different risk factors for the progression of primary biliary cirrhosis". Hepatology. 45 (1): 118–127. doi:10.1002/hep.21472. PMID 17187436. S2CID 19225707.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Nesher G, Margalit R, Ashkenazi YJ (April 2001). "Anti-nuclear envelope antibodies: Clinical associations". Seminars in Arthritis and Rheumatism. 30 (5): 313–320. doi:10.1053/sarh.2001.20266. PMID 11303304.
- ^ Nakanuma Y, Tsuneyama K, Sasaki M, Harada K (August 2000). "Destruction of bile ducts in primary biliary cirrhosis". Baillière's Best Practice & Research. Clinical Gastroenterology. 14 (4): 549–570. doi:10.1053/bega.2000.0103. PMID 10976014.
- ^ a b c d e f g h i j k l m n o p q r s Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M (January 2019). "Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases". Hepatology. 69 (1): 394–419. doi:10.1002/hep.30145. PMID 30070375.
- ^ a b c d Hirschfield GM, Beuers U, Corpechot C, Invernizzi P, Jones D, Marzioni M, et al. (July 2017). "EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis" (PDF). Journal of Hepatology. 67 (1): 145–172. doi:10.1016/j.jhep.2017.03.022. PMID 28427765.
- ^ Levy C, Lindor KD (April 2003). "Treatment Options for Primary Biliary Cirrhosis and Primary Sclerosing Cholangitis". Current Treatment Options in Gastroenterology. 6 (2): 93–103. doi:10.1007/s11938-003-0010-0. PMID 12628068. S2CID 37469838.
- ^ Oo YH, Neuberger J (2004). "Options for treatment of primary biliary cirrhosis". Drugs. 64 (20): 2261–2271. doi:10.2165/00003495-200464200-00001. PMID 15456326. S2CID 1288509.
- ^ Rudic JS, Poropat G, Krstic MN, Bjelakovic G, Gluud C (December 2012). "Ursodeoxycholic acid for primary biliary cirrhosis". The Cochrane Database of Systematic Reviews. 12 (12): CD000551. doi:10.1002/14651858.CD000551.pub3. PMC 7045744. PMID 23235576.
- ^ "Ipsen's Iqirvo receives U.S. FDA accelerated approval as a first-in-class PPAR treatment for primary biliary cholangitis". Ipsen (Press release). 10 June 2024. Retrieved 11 June 2024.
- ^ Hirschfield GM, Shiffman ML, Gulamhusein A, Kowdley KV, Vierling JM, Levy C, et al. (6 April 2023). "Seladelpar efficacy and safety at 3 months in patients with primary biliary cholangitis: ENHANCE, a phase 3, randomized, placebo-controlled study". Hepatology. 78 (2): 397–415. doi:10.1097/HEP.0000000000000395. hdl:11380/1319587. PMC 10344437. PMID 37386786.
- ^ Hasegawa S, Yoneda M, Kurita Y, Nogami A, Honda Y, Hosono K, et al. (1 July 2021). "Cholestatic Liver Disease: Current Treatment Strategies and New Therapeutic Agents". Drugs. 81 (10): 1181–1192. doi:10.1007/s40265-021-01545-7. ISSN 1179-1950. PMC 8282588. PMID 34142342.
- ^ "Gilead's Livdelzi (Seladelpar) Granted Accelerated Approval for Primary Biliary Cholangitis by U.S. FDA" (Press release). Gilead. 14 August 2024. Retrieved 15 August 2024 – via Business Wire.
- ^ Bacon BR, O'Grady JG (2006). Comprehensive Clinical Hepatology. Elsevier Health Sciences. pp. 283–. ISBN 978-0-323-03675-7. Retrieved 29 June 2010.
{{cite book}}: CS1 maint: overridden setting (link) - ^ Collier JD, Ninkovic M, Compston JE (February 2002). "Guidelines on the management of osteoporosis associated with chronic liver disease". Gut. 50 (Suppl 1): i1–i9. doi:10.1136/gut.50.suppl_1.i1. PMC 1867644. PMID 11788576.
- ^ Clavien PA, Killenberg PG (2006). Medical Care of the Liver Transplant Patient: Total Pre-, Intra- and Post-Operative Management. Wiley-Blackwell. p. 155. ISBN 978-1-4051-3032-5.
- ^ Kaneko J, Sugawara Y, Tamura S, Aoki T, Hasegawa K, Yamashiki N, et al. (January 2012). "Long-term outcome of living donor liver transplantation for primary biliary cirrhosis". Transplant International. 25 (1): 7–12. doi:10.1111/j.1432-2277.2011.01336.x. PMID 21923804. S2CID 19872625.
- ^ Carbone M, Mells GF, Pells G, Dawwas MF, Newton JL, Heneghan MA, et al. (March 2013). "Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid". Gastroenterology. 144 (3): 560–569. doi:10.1053/j.gastro.2012.12.005. PMID 23246637.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Lleo A, Wang GQ, Gershwin ME, Hirschfield GM (December 2020). "Primary biliary cholangitis". Lancet. 396 (10266): 1915–1926. doi:10.1016/S0140-6736(20)31607-X. PMID 33308474. S2CID 228086916.
- ^ Cristoferi L, Nardi A, Ronca V, Invernizzi P, Mells G, Carbone M (December 2018). "Prognostic models in primary biliary cholangitis". Journal of Autoimmunity. 95 (1): 171–178. doi:10.1016/j.jaut.2018.10.024. PMID 30420264. S2CID 53292973.
- ^ Carbone M, Nardi A, Flack S, Carpino G, Varvaropoulou N, Gavrila C, et al. (September 2018). "Pretreatment prediction of response to ursodeoxycholic acid in primary biliary cholangitis: development and validation of the UDCA Response Score". The Lancet. Gastroenterology & Hepatology. 3 (9): 626–634. doi:10.1016/S2468-1253(18)30163-8. PMC 6962055. PMID 30017646.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Clavien & Killenberg 2006, p. 429
- ^ Invernizzi P, Miozzo M, Battezzati PM, Bianchi I, Grati FR, Simoni G, et al. (February 2004). "Frequency of monosomy X in women with primary biliary cirrhosis". Lancet. 363 (9408): 533–535. doi:10.1016/S0140-6736(04)15541-4. PMID 14975617. S2CID 5309.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Webb GJ, Ryan RP, Marshall TP, Hirschfield GM (December 2021). "The Epidemiology of UK Autoimmune Liver Disease Varies With Geographic Latitude". Clinical Gastroenterology and Hepatology. 19 (12): 2587–2596. doi:10.1016/j.cgh.2021.01.029. PMC 8661127. PMID 33493696.
- ^ Reuben A (January 2003). "The serology of the Addison-Gull syndrome". Hepatology. 37 (1): 225–228. doi:10.1002/hep.510370134. PMID 12500211. S2CID 42297264.
- ^ Walker JG, Doniach D, Roitt IM, Sherlock S (April 1965). "Serological Tests in Diagnosis of Primary Biliary Cirrhosis". Lancet. 1 (7390): 827–831. doi:10.1016/s0140-6736(65)91372-3. PMID 14263538.
- ^ Mitchison HC, Bassendine MF, Hendrick A, Bennett MK, Bird G, Watson AJ, et al. (1986). "Positive Antimitochondrial Antibody but Normal Alkaline Phosphatase: Is this Primary Biliary Cirrhosis?". Hepatology. 6 (6): 1279–1284. doi:10.1002/hep.1840060609. PMID 3793004. S2CID 13626588.
- ^ Association of Medical Research Charities. "The PBC Foundation". Archived from the original on 4 March 2016. Retrieved 12 July 2015.
- ^ a b Staff (3 January 2008). "Dealing with a silent killer". The Scotsman.
- ^ Thain C (2015). "Primary Biliary Cirrhosis: Getting a Diagnosis". At Home Magazine. Archived from the original (online) on 13 July 2015. Retrieved 28 July 2015.
- ^ Jacoby A, Rannard A, Buck D, Bhala N, Newton JL, James OF, et al. (November 2005). "Development, Validation, and Evaluation of the PBC-40, a Disease Specific Health-Related Quality of Life Measure for Primary Biliary Cirrhosis". Gut. 54 (11): 1622–1629. doi:10.1136/gut.2005.065862. PMC 1774759. PMID 15961522.
- ^ Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, et al. (March 2011). "Genome-Wide Association study Identifies 12 New Susceptibility Loci for Primary Biliary Cirrhosis". Nature Genetics. 43 (4): 329–332. doi:10.1038/ng.789. PMC 3071550. PMID 21399635.
{{cite journal}}: CS1 maint: overridden setting (link) - ^ Gordon B (31 December 2003). "A royal seal of approval". The Scotsman.
- ^ PBC Foundation. "EASL Name Change Presentation". Archived from the original on 9 July 2015. Retrieved 8 July 2015.
- ^ Kim M (18 January 2015). "New hope for PBC liver disease". ABC30 Action news. Retrieved 4 August 2015.