
Introduction
editmicroRNA sequencing (miRNA-seq) was developed as an application of next-generation sequencing or massively parallel high-throughput sequencing technologies to uncover novel miRNAs and their expression profiles in a given sample.
MicroRNAs (miRNAs) family of small ribonucleic acids, 21-25 nucleotides in length, that modulate protein expression through inhibition of translation 1-3 [1][2][3]. The first miRNA, lin-4, was discovered in C eleans in a genetic mutagenesis screen looking to identify molecular elements that control post-embryonic development [4]. Additional studies on the lin-4 gene revealed it encoded a 22nt non-coding RNA with conserved complementary binding sites in the 3’-untranslated region of the lin-14 mRNA transcript [5]. The location of the binding sites, tied in with the fact lin-4 downregulated LIN-14 protein expression [6] suggested lin-4 was inhibiting the translation of lin-14. Continued research into miRNA mediated gene regulation revealed miRNAs are involved in the regulation of many developmental and biological processes including haematopoiesis (miR-181 in Mus musculus[7]), lipid metabolism (miR-14 in Drosophila melanogaster[8]) and neuronal development (lsy-6 in Caenorhabditis elegans [9])[10]. The uncovering of miRNAs as a fundamental cellular regulatory mechanism then led to rapid development of different techniques to identify and characterize miRNAs such as miRNA-SEQ.
History
editmiRNA sequencing in of itself is not a novel idea, initial methods of sequencing utilized Sanger sequencing methods. Sequencing preparation involved creating libraries by cloning of DNA reverse transcribed from endogenous small RNAs of 21–25 bp size selected by column and gel electrophoresis[11]. However, this method is exhaustive in turns of time and resources, as each clone has to be individually amplified and prepared for sequencing. This method also inadvertently favors miRNAs that are highly expressed[12]. Next-generation sequencing eliminates the need for sequence specific hybridization probes required in microarray analysis as well as laborious cloning methods required in the Sanger sequencing method. Additionally, next-generation sequencing platforms in the miRNA-SEQ method facilitate the sequencing of large pools of small RNAs in a single sequencing run[13].
miRNA-SEQ can be performed using a verity of sequencing platforms. The first analysis of small RNAs using miRNA-SEQ methods examined approximately 400,000 small RNAs from Caenorhabditis elegans using Life Sciences 454 Sequencing platform. This study identified 18 novel miRNA genes as well as a new class of nematode small RNAs termed 21U-RNAs[14]. Another study comparing small RNA profiles of human cervical tumours and normal tissue, utilized the Illumina Genome Analyzer to identify 64 novel human miRNA genes as well as 67 differentially expressed miRNAs[15]. Applied Biosystems SOLiD sequencing platform as also been used to examine the prognostic value of miRNAs in detecting human brest cancer[16].
Methods
editSmall RNA Preparation
edit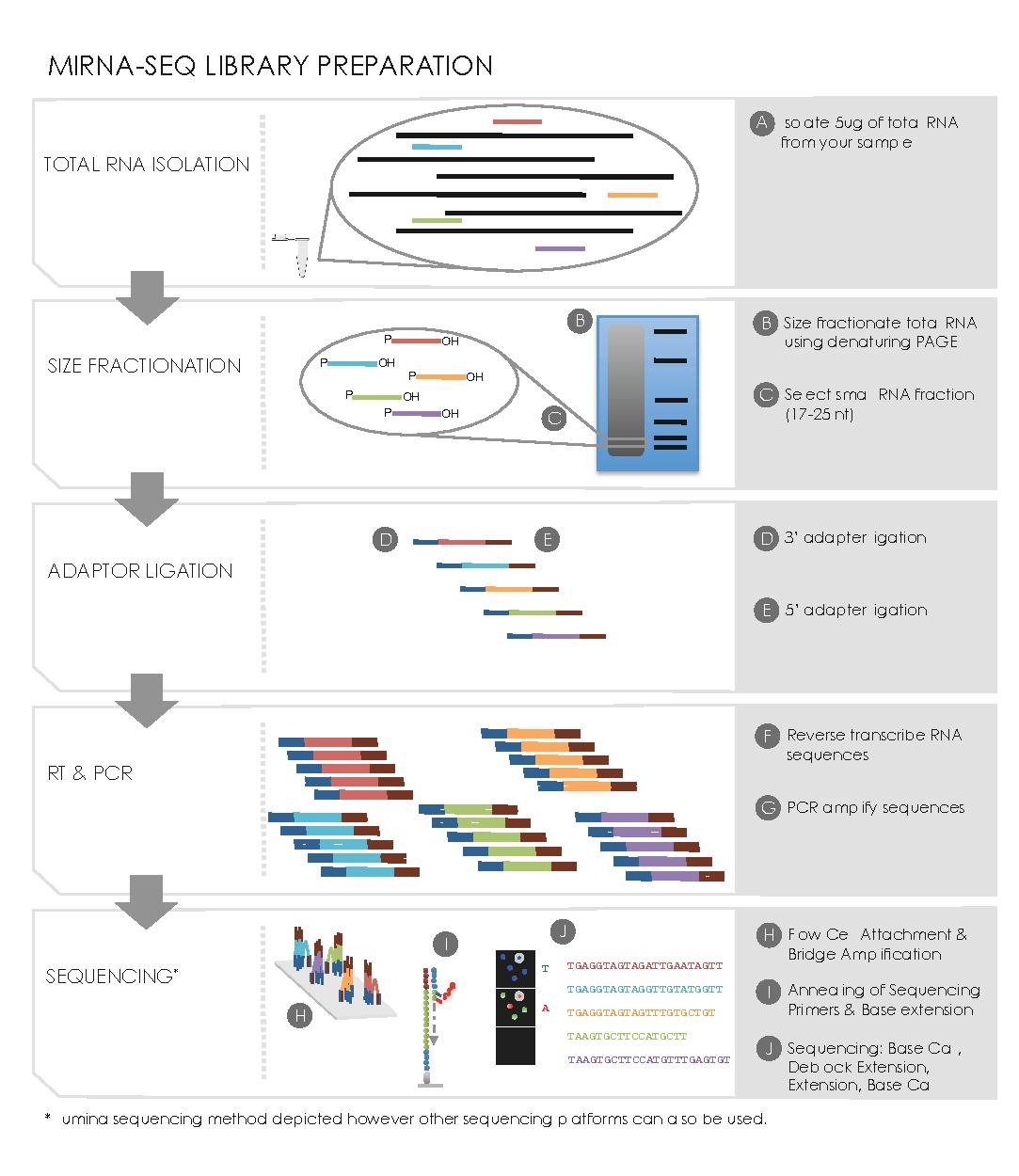
Sequencing
editData Analysis
editCentral to miRNA-seq data analysis is the ability to 1) obtain miRNA abundance levels from sequence reads, 2) discover novel miRNAs and then be able to 3) determine the differentially expressed miRNA and their 4) associated mRNA gene targets.
miRNA Alignment & Abundance Quantification
editmiRNAs may be preferentially expressed in certain cell types, tissues, stages of development, or in particular disease states such as cancer[17]. Since deep sequencing (miRNA-seq) generates millions of reads from a given sample, it allows us to profile miRNAs; whether it may be by quantifying their absolute abundance, to discover their variants (known as isomirs[18]) Note that given that the average length of sequence reads are longer than the average miRNA (17-25 nt), the 3’ and 5’ ends of the miRNA should be found on the same read. There are several miRNA abundance quantification algorithms [19][20]. Their general steps are as follows[21] :
- After sequencing, the raw sequence reads are filtered based on quality. The adaptor sequences are also trimmed of the raw sequence reads.
- The resulting reads are then formatted into a fasta file where the copy number and sequence is recorded for each unique tag.
- Sequences that may represent E. Coli contamination are identified by a BLAST search against an E. Coli database and removed are from analysis.
- Each of the remaining sequences are aligned against a miRNA sequence database (such as miRBase[22]) In order to account for imperfect DICER processing, a 6nt overhang on the 3’ end, and 3nt on the 5’ end are allowed.
- The reads that do not align to the miRNA database are then loosely aligned to miRNA precursors to detect miRNAs that might carry mutations or those that have gone through RNA editing.
- The read counts for each miRNA are then normalized to the total number of mapped miRNAs to report the abundance of each miRNA.
Novel miRNA Discovery
editAnother advantage of miRNA-seq is that it allows the discovery of novel miRNAs that may have eluded traditional screening and profiling methods. [23] There are several novel miRNA discovery algorithms. Their general steps are as follows:
- Obtain reads that did not align to known miRNA sequences, and map them to the genome.
- RNA Folding Method
- For the miRNA sequences were an exact match is found, obtain the genomic sequence including ~100bp of flanking sequence on either side, and run the RNA through RNA folding software such as the Vienna package[24].
- Folded sequences that lie on one arm of the miRNA hairpin and have a minimum free energy of less than ~25kcal/mol are shortlisted as putative miRNA.
- The shortlisted sequences are trimmed down to include only the possible precursor sequence and are then refolded to ensure that the precursor was not artificially stabilized by neighbouring sequences.
- The resulting folded sequences are considered novel miRNAs if the miRNA sequence falls within one arm of the hairpin, and are highly conserved between species.
- Star Strand Expression Method (miRdeep[25])
- Novel miRNA sequences are identified based on the characteristic expression pattern that they display due to DICER processing: higher expression of the mature miRNA over the star strand and loop sequences.
Differential Expression Analysis
editAfter the abundances of miRNAs are quantified for each sample, their expression levels can be compared between samples. One would then be able to identify miRNA that are preferentially expressed that particular time points, or in particular tissues or disease states. After normalizing the for number of mapped reads between samples, one can use a host of statistical tests (like those used in gene expression profiling) to determine differential expression
Target Prediction
editIdentifying a miRNA’s mRNA targets will provide an understanding of the genes or networks of genes whose expression they regulate [26]. Public databases provide predictions of miRNA targets. But to better distinguish true positive predictions from false positive predictions, miRNA-seq data can be integrated to mRNA-seq data to observe for miRNA:mRNA functional pairs. TargetScan[27], miRanda[28], and PicTar[29] are software designed for this purpose. A list of prediction software is given [here] The general steps are:
- To determine miRNA:mRNA binding pairs, complementarity between the miRNA sequences at the 3’-UTR of the mRNA sequence is identified. Typically one more bp mismatches are allowed since miRNA binding is not perfectly complementary.
- The degree of conservation of miRNA:mRNA binding pairs across species is determined. Typically, more highly binding pairs are less likely to be false positives of prediction.
- Observe for evidence of miRNA targeting in mRNA-seq or protein expression data: where the miRNA expression is high, the gene and protein expression of its target gene should be low.
Applications
editIdentification of Novel miRNAs
editmiRNA-seq has revealed novel miRNAs that were previously eluded in traditional miRNA profiling methods. Examples of such findings are in embryonic stem cells[30], chicken embryos[31], acute lymphoblastic leukemia[32], diffuse large b-cell lymphoma and b-cells[33], acute myeloid leukemia[34], and lung cancer[35].
Disease Biomarks
editComparison With Other Methods of miRNA Profiling
editThe disadvantage of using miRNA-seq over other methods of miRNA profiling is that it is more expensive, requires a larger amount of total RNA, and is more time consuming than microarray and qPCR methods[36]. As well, miRNA-seq library preparation methods seem to have systematic preferential representation of the miRNA complement, and thisprevents accurate determination of miRNA abundance[37]. Despite these disadvantages, miRNA-seq remains the platform of choice for profiling miRNA. The approach is hybridization independent and therefore does not require a priori information. As such, its advantages over previous miRNA profiling techniques include allowing one to see different miRNA isoforms (isoMirs) or very similar miRNAs and allows the identification of point mutations in miRNA genes. miRNA-seq also allows for the validation of novel miRNA discovery and predictions at the nucleotide level [38].
References
edit- ^ Kim, V. N., Han, J. & Siomi, M. C. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10, 126–139 (2009).
- ^ Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004).
- ^ He, L. & Hannon, G. J. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5, 522–531 (2004).
- ^ Ambros, V. A hierarchy of regulatory genes controls a larva-to-adult developmental switch in Caenorhabditis elegans. Cell 57, 49–57 (1989).
- ^ Lee, R. C., Feinbaum, R. L. & Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854 (1993).
- ^ Wightman, B., Ha, I. & Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855–862 (1993).
- ^ Chen, C.-Z., Li, L., Lodish, H. F. & Bartel, D. P. MicroRNAs modulate hematopoietic lineage differentiation. Science 303, 83–86 (2004).
- ^ Xu, P., Vernooy, S. Y., Guo, M. & Hay, B. A. The Drosophila microRNA Mir-14 suppresses cell death and is required for normal fat metabolism. Curr. Biol. 13, 790–795 (2003).
- ^ Johnston, R. J. & Hobert, O. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature 426, 845–849 (2003).
- ^ He, L. & Hannon, G. J. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5, 522–531 (2004).
- ^ Lee, R. C. & Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 294, 862–864 (2001).
- ^ Chen, C.-Z., Li, L., Lodish, H. F. & Bartel, D. P. MicroRNAs modulate hematopoietic lineage differentiation. Science 303, 83–86 (2004).
- ^ Aldridge, S. & Hadfield, J. Introduction to miRNA profiling technologies and cross-platform comparison. Methods Mol. Biol. 822, 19–31 (2012).
- ^ Ruby, J. G. et al. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell 127, 1193–1207 (2006).
- ^ Witten, D., Tibshirani, R., Gu, S. G., Fire, A. & Lui, W.-O. Ultra-high throughput sequencing-based small RNA discovery and discrete statistical biomarker analysis in a collection of cervical tumours and matched controls. BMC Biol. 8, 58 (2010).
- ^ Wu, Q. et al. Next-generation sequencing of microRNAs for breast cancer detection. J. Biomed. Biotechnol. 2011, 597145 (2011).
- ^ Farazi, T. A., Spitzer, J. I., Morozov, P. & Tuschl, T. miRNAs in human cancer. J Pathol 223, 102–115 (2011).
- ^ Morin, R. D. et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res 18, 610–621 (2008).
- ^ Shendure, J. & Ji, H. Next-generation DNA sequencing. Nat Biotechnol 26, 1135–1145 (2008).
- ^ Berninger, P., Gaidatzis, D., van Nimwegen, E. & Zavolan, M. Computational analysis of small RNA cloning data. Methods 44, 13–21 (2008).
- ^ Creighton, C. J., Reid, J. G. & Gunaratne, P. H. Expression profiling of microRNAs by deep sequencing. Brief Bioinformatics 10, 490–497 (2009).
- ^ Kozomara, A. & Griffiths-Jones, S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39, D152–7 (2011).
- ^ Creighton, C. J., Reid, J. G. & Gunaratne, P. H. Expression profiling of microRNAs by deep sequencing. Brief Bioinformatics 10, 490–497 (2009).
- ^ Hofacker, I., Fontana, W., Stadler, P. & Bonhoeffer, L. Fast folding and comparison of RNA secondary structures (the Vienna RNA Package) Chemical Monthly. 1994; 125: 167–188. doi: 10.1007. (9999).
- ^ Yang, X. & Li, L. miRDeep-P: a computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 27, 2614–2615 (2011).
- ^ Cloonan, N. et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol 12, R126 (2011).
- ^ Garcia, D. M. et al. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol 18, 1139–1146 (2011).
- ^ Mazière, P. & Enright, A. J. Prediction of microRNA targets. Drug Discov. Today 12, 452–458 (2007).
- ^ Krek, A. Identification of microRNA targets. DAI-B 70/07, (2010).
- ^ Morin, R. D. et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res 18, 610–621 (2008).
- ^ Buermans, H. P. J., Ariyurek, Y., van Ommen, G., Dunnen, den, J. T. & 't Hoen, P. A. C. New methods for next generation sequencing based microRNA expression profiling. BMC Genomics 11, 716 (2010).
- ^ Zhang, H. et al. Genome-wide analysis of small RNA and novel MicroRNA discovery in human acute lymphoblastic leukemia based on extensive sequencing approach. PLoS ONE 4, e6849 (2009).
- ^ Jima, D. D. et al. Deep sequencing of the small RNA transcriptome of normal and malignant human B cells identifies hundreds of novel microRNAs. Blood 116, e118–27 (2010).
- ^ Starczynowski, D. T. et al. Genome-wide identification of human microRNAs located in leukemia-associated genomic alterations. Blood 117, 595–607 (2011).
- ^ Keller, A. et al. Next-generation sequencing identifies novel microRNAs in peripheral blood of lung cancer patients. Mol Biosyst 7, 3187–3199 (2011).
- ^ Baker, M. MicroRNA profiling: separating signal from noise. Nat Methods 7, 687
- ^ Linsen, S. E. V. et al. Limitations and possibilities of small RNA digital gene expression profiling. Nat Methods 6, 474–476 (2009).
- ^ Git, A. et al. Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 16, 991–1006 (2010).