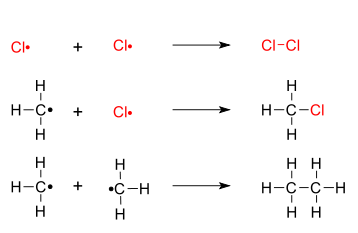
A radical abstracts a hydrogen atom from methane, leaving a primary methyl radical. The methyl radical then abstracts Cl• from Cl2 to give the desired product and another chlorine radical. Methane chlorination: propagation The radical will then participate in another propagation reaction: the radical chain. Other products such as CH2Cl2 may also form.
Two free radicals (chlorine and chlorine, chlorine and methyl, or methyl and methyl) combine: Methane chlorination: termination The last possibility generates in an impurity in the final mixture (notably, an organic molecule with a longer carbon chain than the reactants).
Radical fluorination with the pure element is difficult to control and highly exothermic; care must be taken to prevent an explosion or a runaway reaction. With chlorine the reaction is moderate to fast; with bromine, slow and requires intense UV irradiation; and with iodine, it is practically nonexistent and thermodynamically unfavored. However, radical iodination can be completed with other iodine sources (see § Variants).[2]
The different rates are often a pedagogical demonstration of the reactivity–selectivity principle and the Hammond postulate. A bromine radical is not very reactive and the transition state for hydrogen abstraction has much radical character and is reached late. The reactive chlorine radical develops a transition state resembling the reactant with little radical character. When the alkyl radical is fully formed in the transition state, it can benefit fully from any resonance stabilization present thereby maximizing selectivity.[dubious – discuss][citation needed]
Aside from those few exceptions, free-radical halogenation is notoriously unselective. Chlorination rarely stops at monosubstitution:[2] depending on reaction conditions, methane chlorination yields varying proportions of chloromethane, dichloromethane, chloroform and carbon tetrachloride.
For asymmetric substrates, the reaction produces all possible isomers, but not equally. Radical halogenations are generally indifferent amongst equi-substituted potential radicals and effect a so-called statistical product distribution. Butane (CH3−CH2−CH2−CH3), for example, can be chlorinated at the "1" position to give 1-chlorobutane (CH3−CH2−CH2−CH2Cl) or at the "2" position to give 2-chlorobutane (CH3−CH2−CHCl−CH3). The latter occurs faster, and is the major product.
Thus any single chlorination step slightly favors substitution at the carbon already most substituted. The rates are generally constant across reactions and predict product distributions with relatively high accuracy.[3][4] For example, 2-methyl butane ((CH3)2CHCH2CH3) exhibits the following results:
Moiety
Type
# Hydrogens
Relative rate
Quantity
Proportion
2CH3
Primary
6
×
1
=
6
28%
CH
Tertiary
1
×
5
=
5
23%
CH2
Secondary
2
×
3.8
=
7.6
35%
CH3
Primary
3
×
1
=
3
14%
Total
21.6
100%
Note that the sole tertiary hydrogen is nearly as likely to chlorinate as the 6 hydrogens terminating the branches, despite their much greater abundance.